BemSn (m,n=1-3) Clusters to Hexagonal BeS Material: A Density Functional Study
Majeed Shaikh, Vipin Kumar and Debesh R. Roy
Majeed Shaikh, Vipin Kumar and Debesh R. Roy*
Department of Applied Physics, S. V. National Institute of Technology, Surat 395007, India
- *Corresponding Author:
- Debesh R. Roy
Department of Applied Physics, S. V. National Institute of Technology, Surat 395007, India
Tel: +91-(261) 2201542
Fax: +91 (261) 2227334
E-mail: drr.iit@gmail.com
Received date: October 15, 2015, Accepted date: December 03, 2015, Published date: December 10, 2015
Citation: Roy DR. BemSn (m,n=1-3) Clusters to Hexagonal BeS Material: A Density Functional Study. Nano Res Appl. 2016, 2:1.
Abstract
A detailed investigation on the structure, electronic and thermodynamic properties for a series of group II and VI Combined hybrid alloy clusters, viz, BemSn (m,n=1-3) is performed, under density functional theory (DFT). The geometries of all clusters has been optimized by employing a very popular and reliable exchange-correlation functional, viz, Becke’s 3 parameter exchange with Lee-Yang-Parr correlation functional (B3LYP). The influence of adding of group II and/or group VI element on the electronic properties is also addressed in the present work. A sincere effort has been tendered to identify any potential cluster motif from the considered series for utilizing as the building block for prospective metal-insulator-semiconductor (MIS) materials at the bulk scale. Accordingly, we have studied we have studied hexagonal barium sulfide (BeS) material at bulk scale. The band structure and projected density of states for the bulk BeS indicates its metallic nature.
Keywords
Binary clusters; Jellium model; Barium sulfide; Density functional theory
Introduction
The journey of utilizing atomic and molecular clusters for designing novel nanomaterials [1-4] has been initiated with the discovery of fullerene [5]. The effect of quantum confinement [6] often show dramatic changes in the physicochemical properties of the materials at the sub-nano or nano-scale are compared to their bulk counterparts. This unusual behavior of nano-scale materials is found to be very useful in various kinds of applications to the mankind for the last three decades [1-4]. For cluster or nano-scale materials, the tendency of coalesce due to their metastable nature in many cases, is a difficulty for their practical use. Along this line, a number of techniques are developed to overcome this situation, e.g., isolation of clusters by passivating them with inorganic [7] or organic ligands [8], inserting them into cages (zeolite) [1] or depositing them on surfaces [9]. On the way for developing novel materials, one of the important aspects to the researchers is to predict or identify clusters with unusual or exceptional stability or useful physicochemical properties, which may therefore be utilized as the building block for modeling and synthesize of useful materials at the nano or bulk scale with tunable chemical, electronic, optical and magnetic properties [4,6].
It is known that, the materials originated from the combinations of groups III and V, II and VI elements often exhibit important properties and usefulness [1-4,6]. Recently, small beryllium clusters and their alloys with other elements have gained lots of attention due to their interesting properties and possible applications [10-13]. Electronic structure of beryllium clusters is investigated by Cerowski et al. [10]. Chattaraj and Roy [11,12] have reported exceptional stability of the Be32- cluster due to its π aromaticity, and demonstrated various stable sandwich complexes of Be32-. Very recently, Yu and Gao [13] have studied 2D hexagonal beryllium sulfide (h-BeS) sheet. They have found h-BeS shows an indirect energy gap of 4.26 eV and thermodynamic stability up to 1000 K [13].
The purpose of the present investigation is to predict geometries, electronic properties and search for any unusual or exceptional stable cluster building motifs from a series of heteroatomic (group II and group VI) clusters, BemSn (m,n=1-3). The stability of these clusters is assessed with considering various physicochemical properties, e.g., energy gain in adding a Be or S atom (ΔEBe or ES) to the previous sizes, highest occupied and lowest unoccupied molecular orbitals energy gap (HLG), ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical hardness (η) etc., and explained under jellium framework. Also, identified useful cluster unit is utilized to investigate the structure and electronic properties of bulk hexagonal BeS (bulk h-BeS) material at the bulk scale. The band structure and projected density of states (PDOS) for the newly reported hexagonal BeS material is reported in the present paper.
Theory and Computation
BemSn (m,n=1-3) Clusters
The theoretical investigations are carried out using a gradient corrected approach within the density functional theory (DFT) framework [14-16]. A molecular orbital approach, using a linear combination of atomic orbitals, is applied to probe electronic structure. For exchange and correlation functional, we have used a very popular and successive hybrid functional, Becke 3-parameter exchange and Lee-Yang-Parr correlation (B3LYP) which has been introduced by Becke in 1993 [17]. We have used LANL2DZ (Los Alamos ECP plus DZ) basis set which includes a scalar relativistic correction [18,19].
The energy gain (ΔES) in forming BemSn clusters by adding a sulphur atom (for fixed Be atom) to the preceding BemSn-1 (m=1 or 2 or 3; n=1-3) size is giving as,
ΔES=E(S)+E(BemSn-1)–E(BemSn) (1)
where E(BenSn), E(BemSn-1) and E(S) are the total energies of the BenSn, BemSn-1 clusters and of the S atom, respectively.
The ionization potential (IP) and electron affinity (EA) can be expressed in terms of the highest occupied (?HOMO) and the lowest unoccupied (?LUMO) molecular orbital energies using Koopmans’ approximation [16] as,
IP ≈–?HOMO ; EA ≈–?LUMO (2)
To account for the stability of a molecule, Pearson [20] has introduced ‘chemical hardness’. For an N-electron system, the second derivative of energy with respect to N, keeping external potential  fixed, is considered to be a measure of the chemical hardness [21]:
fixed, is considered to be a measure of the chemical hardness [21]:
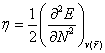 (3)
(3)
The hardness can be expressed in terms of ?HOMO and ?LUMO as follows [16]:
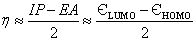 (4)
(4)
h-BeS Bulk Material
To investigate the structure and electronic properties of hexagonal bulk 3D BeS, first principle calculations are performed based on fully self-consistent density functional theory (DFT) [14-16] with the plane wave pseudopotential codes as implemented in the QUANTUM-ESPRESSO package [22]. In all calculations, the exchange-correlation effects have been treated within the Perdew-Burke-Ernzerhof (PBE) [23]. We have used the PAW pseudo-potential for Be and S atom along with a plane wave basis with a wave-function kinetic-energy cut off 60 Ry and a cutoff of 600 Ry for the charge density. Hexagonal 2D planar structure is considered with an S atom at (0, 0, 0) and the Be atom at (1/3, 2/3, 0) & (2/3, 1/3, 0). We have used 25 × 25 × 25 Å for bulk Monkhorst pack k-mesh along the path Γ-K-M-Γ in the irreducible Brillouin zone (IBZ) to obtain the band structure with very fine mesh point. The convergence threshold scale for self-consistency cycles was 10-10 Ry. All atomic positions and cell parameters were fully relaxed with a tolerance of 10-3 Ry. Crystal structure was visualized and drawn using XCRYSDEN software [24].
Results and Discussion
Figure 1 represents the ground state geometries of BemSn (m, n=1- 3) hybrid alloy clusters along with their respective geometrical parameters. The bond lengths varies for the different combinations are as follows: Be-Be (1.97-2.53 Å), Be-S (1.79-1.98 Å) and S-S (2.42-2.65 Å). Table 1 shows various electronic properties of BemSn (where m, n=1-3) clusters, viz., HOMO-LUMO energy gap (HLG), ionization potential (I.P.), electron affinity (E.A.), chemical hardness (η), electronegativity (χ) and energy gain for adding one S atom to the previous size (ΔES). It may be noted that the HLG value for the selected nine clusters ranges between 2.3 eV to 4.84 eV, implying an initial expectation for the beryllium sulphide materials at bulk scale among the semiconductor-insulator region in MIS arena. The higher I. P. (>5 eV, except for BeS2) and larger chemical hardness (η) (>1 eV) implies the stability of the selected clusters in general.
| m,n | HLG (eV) |
I.P. (eV) |
E.A. (eV) |
η (eV) |
χ (eV) |
ΔES (eV) |
|---|---|---|---|---|---|---|
| 1,1 | 2.33 | 6.73 | 4.40 | 1.16 | 5.56 | 4.56 |
| 1,2 | 2.30 | 3.62 | 2.32 | 1.15 | 5.47 | 2.10 |
| 1,3 | 2.86 | 6.98 | 4.11 | 1.43 | 5.54 | 1.86 |
| 2,1 | 2.72 | 5.77 | 3.06 | 1.36 | 4.41 | 2.10 |
| 2,2 | 3.34 | 6.77 | 3.42 | 1.67 | 5.09 | 5.21 |
| 2,3 | 3.11 | 7.02 | 3.91 | 1.56 | 5.46 | 5.12 |
| 3,1 | 2.64 | 5.23 | 2.59 | 1.32 | 3.91 | 1.86 |
| 3,2 | 3.95 | 6.80 | 2.86 | 1.97 | 4.83 | 2.38 |
| 3,3 | 4.84 | 7.67 | 2.83 | 2.42 | 3.25 | 5.67 |
Table 1: HOMO-LUMO energy gap (HLG), ionization potential (I.P.), electron affinity (E.A.), chemical hardness (η), electronegativity (χ) and energy gain in adding a S atom (ΔES) to an existing BemSn-1 (m=1 or 2 or 3; n=1-3), of the BemSn (m, n=1-3) clusters.

Figure 1: Ground state structures and associated point groups of BemSn (m, n=1-3) clusters.
Figure 2 shows the energy gain (ES) of BemSn (where m, n=1-3) for adding one S atom to the previous size by keeping beryllium size fixed (m=1, 2 or 3), as shown on Figure 2a, Figure 2b and Figure 2c respectively. It may be noted that the maximum ΔES for all three series are 4.56 eV (for BeS, Figure 2a), 5.21 eV (for Be2S2, Figure 2b) and 5.67 eV (for Be3S3, Figure 2c) respectively. It is heartening to note that the clusters with equal Be and S stoichiometry (m:n=1:1, 2:2, 3:3) shows maximum energy gain in the respective series, identifying them as unusual stable compared to their neighbour. Therefore an equal stichiometry for beryllium and sulphur for developing novel berrylium sulfide material both at nano and bulk scale may be considered as the best combination.

Figure 2: Energy gain (ΔES) of BemSn (m, n=1-3) clusters: (a) BemSn (m=1, n=1-3), (b) BemSn (m=2, n=1-3) and (c) BemSn (m=3, n=1-3).
Along this direction, we have explored the possibility for developing a novel hexagonal beryllium sulphide (h-BeS) bulk material by considering one of the exceptionally stable rhombus shaped cluster Be2S2 as the building block. The D2h symmetry of Be2S2 makes it possible for considering as the building unit for our desired hexagonal beryllium sulphide (h-BeS) bulk material. Figure 3 represents the optimized unit cell (Figure 3a) and bulk h-BeS (Figure 3b) along with their geometrical parameters. Optimized Be-S bond distance is found as 2.31 Å.

Figure 3 (a) Unit cell and (b) Minimized geometry of the bulk h-BeS.
The calculated band structure along with the projected density of states (PDOS) for bulk h-BeS is shown in Figure 4. The band structure is plotted along the high symmetry points, Γ-K-M-Γ of the irreducible Brillouin zone (IBZ). From the predicted PDOS plot, it is clear that in the valance band, main from 2p-orbitals of both the Be and S, dominated with 2p-orbital 2p-orbital of S. The overlapping feature of bands at the Fermi level clearly indicates the metallic nature of our developed bulk h-BeS. It may be noted that Yu and Gao [13] have found 2D h-BeS as insulator (indirect band gap of 4.26 eV). Our investigation indicates that change of materials dimension (2D to bulk 3D) dramatically change the nature of the h-BeS material from insulator to metallic! This outcome deserves further future investigation on beryllium sulfide for its potential applications.

Figure 4: Electronic band structure and projected density of states (PDOS) of bulk h-BeS. The PDOS is resolved into s and p components. The total DOS is also shown.
Conclusions
A detail first principle investigation is performed in search of the exceptional stable clusters in the group II and VI combined hybrid alloy clusters BemSn (m, n=1-3) series. The calculation of various electronic properties, e.g., HOMO-LUMO energy gap (HLG), ionization potential (I.P.), electron affinity (E.A.), chemical hardness (η) etc. shows that the selected clusters are stable in general. The energy gain (ΔES) calculation shows that BemSn clusters (BeS, Be2S2 and Be3S3) with equal stoichiometric ratios for Be and S (m=n) are unusually stable compared to their neighbors. To investigate the possible metal-insulator-semiconductor (MIS) behavior of these clusters at the bulk scale, we have chosen one of the exceptionally stable clusters Be2S2 as the building block for bulk hexagonal BeS (h-BeS) material. The rhombus shaped D2h symmetric Be2S2 makes it suitable as the unit for hexagonal bulk morphology. The optimized Be-S distance is found to be 2.31 Å for bulk h-BeS. The projected density of states (PDOS) shows that p orbitals of sulphur plays major contribution in the valance band. The overlapping of the energy bands at the Fermi level as noticed in the predicted band structure for bulk h-BeS clearly implies the metallic nature of our developed material. It also be noted that the dramatic change in to metallic behavior for our developed bulk h-BeS (3D) from the previously reported insulator 2D h-BeS by the researchers demands a critical future investigation on beryllium sulfide materials for potential applications in various industrial fields.
Acknowledgment
DRR is thankful to Department of Science & Technology, Govt. of India for financial support (Sanc. No.SR/FTP/PS-199/2011).
References
- Bein TH, Jacobs PA, Schmidt F (1982) Metal Microclusters in Zeolites, edited by PA. Jacobs Elsevier, Amsterdam.
- KrügerA (2010) Carbon Materials and Nanotechnology Wiley-VCH, NY.
- Handbook of nanotechnology (3rdedn), Ed. B. Bhushan (Springer-Verlag, NY, 2010).
- Nanoclusters: A Bridge Across Disciplines, Eds. P. Jena and A.W. Castleman, Jr. (Elsevier, Amsterdam, 2010).
- Kroto HW, Heath J, O'Brien SC, Curl RF, Smalley RE (1985) Nature London 318: 162.
- Quantum Phenomena in Clusters and Nanostructures, Eds. S.N. Khanna and A.W. Castleman, Jr. (Springer, New York, 2003).
- Claridge SA, CastlemanJr AW, Khanna SN, Murray CB, Sen A, et al.(2009) ACS Nano 3: 244.
- Zou R, Abdel-FattahAI, XuH, Zhao Y,Hickmott DD (2010)CrystEngComm 12: 1337.
- Brane H, Giovannini M, Bromann K, Kern K (1998) Nature 394: 451.
- Cerowski V, Rao BK, Khanna SN, Jena P, Ishii S, et al. (2005) Evolution of the electronic structure of Be clusters. J Chem Phys 123: 074329.
- Chattaraj PK, Roy DR (2007) Aromaticity in polyacene analogues of inorganic ring compounds. J PhysChemA 111: 4684-4696.
- Chattaraj PK, Roy DR, Duley S (2008) Chem Phys Lett 460: 382.
- Yu J, Guo W (2013) Two-Dimensional Hexagonal Beryllium Sulfide Crystal. J Phys Chem Lett 4: 1856-1860.
- Hohenberg P,Kohn W (1964) Phys Rev B 136: B864.
- Kohn W,Sham LJ (1965) Phys Rev A 140: A1133.
- Parr RG,Yang W (1989) Density Functional Theory of Atoms and Molecules Oxford University Press, New York.
- Becke AD (1993) J Chem Phys 98: 5648.
- Dunning JrTH, Hay PJ (1976) in Modern Theoretical Chemistry, Ed. H. F. Schaefer III, Vol. 3 Plenum, New York, pp. 1-28.
- Hay PJ,Wadt WR (1985) J Chem Phys 82: 270.
- Pearson RG (1973) Hard and Soft Acids and Bases Dowden, Hutchinson and Ross, Stroudsberg, PA.
- Parr RG,PearsonRG (1983) J Am ChemSoc 105: 7512.
- Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, et al. (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J PhysCondens Matter 21: 395502.
- Perdew JP, Burke K, Ernzerhof M (1996)Phys Rev Lett 77: 3865.
- Kokalj A (2003) Comp Mater Sci 28: 155.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences